RPS Case of the Month
Dec 2023
Unexpected finding in 65-year-old male with chronic kidney disease and minimal proteinuria
Siarhei Dzedzik1
Ilya Glezerman1,2
Surya V. Seshan1
1. New York-Presbyterian/Weill Cornell Medicine, New York, NY2. Memorial Sloan Kettering Cancer Center, New York, NY
CLINICAL HISTORY
65-year-old male who initially presented to the local hospital after a fall. An imaging study revealed an osteolytic lesion with suspected pathological fracture of C6 vertebral body. Following resection of the lesion, the patient received radiation to the affected area. Pathological examination showed tumor cells positive for CD138 with kappa restriction, consistent with the diagnosis of plasmacytoma. Two and half years later, he was evaluated for elevated creatinine, which increased from a baseline of 1.0 (N = 0.6-1.3) mg/dl to 1.5 mg/dL. The physical exam was unremarkable. Comprehensive metabolic panel was also unremarkable. In addition, there was an elevation of free kappa light chains to 38.09 (0.33-1.94) mg/dL, kappa to lambda ratio of 35.93 (0.26-1.65), serum protein electrophoresis was negative for M-spike and blood immunofixation was positive for IgA kappa monoclonal gammopathy. The urinary protein electrophoresis was negative for M-spike but urine immunofixation was positive for monoclonal free kappa light chain band. Urinary protein-to-creatinine ratio (UPC) was less than 0.1 and urinalysis was devoid of cells and sediment. Initially patient was followed conservatively but underwent a kidney biopsy ten months after presentation to the renal clinic because of rising free kappa light chains (84.07 mg/dL) and development of proteinuria with a UPC 0.6.
BIOPSY FINDINGS
Light microscopy revealed mild ischemic changes in nine out of forty-seven glomeruli (9/47). The remaining glomeruli were unremarkable. Nearly 30% of the cortical tissue showed tubular atrophy with mild interstitial fibrosis and minimal to sparse chronic interstitial inflammatory infiltrates, composed of lymphocytes admixed with scattered macrophages and occasional plasma cells. Tubular findings are summarized in Figure 1.
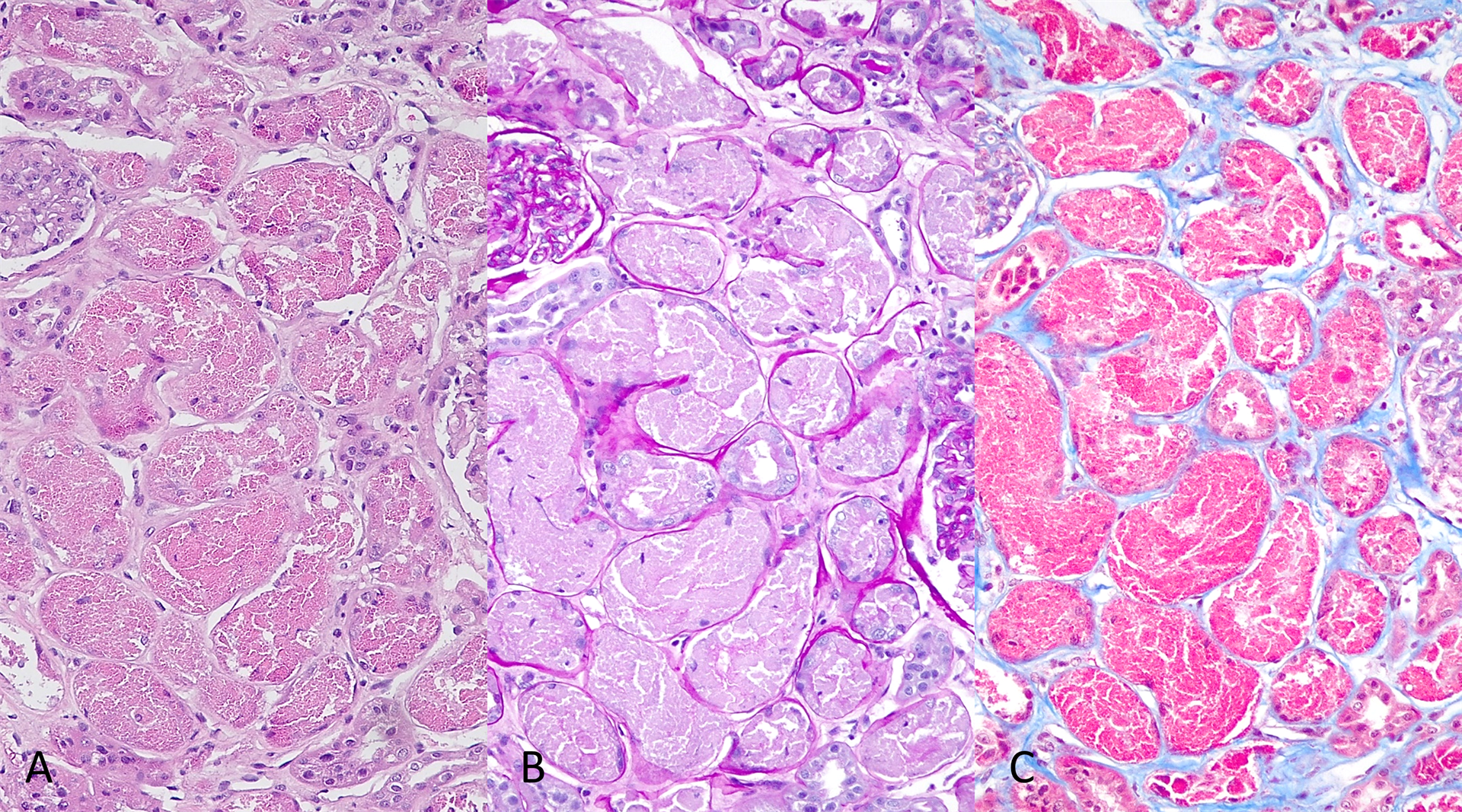
Figure 1: Most of the proximal tubules appear enlarged. The epithelial cells are distended by coarsely granular, eosinophilic material (A) that is PAS-negative (B) and strongly fuchsinophilic on trichrome stain (C). There is an accompanying tubular epithelial injury that ranges from the loss of brush border, karyopyknosis to the loss of cellular cohesion and their subsequent sloughing into the lumen.

IMMUNOFLUORESCENCE MICROSCOPY (IF):
Immunofluorescence microscopy (2 glomeruli), performed on frozen tissue (IF-F), revealed no staining for immunoglobulins IgG, IgA, complement components C3, C1q, kappa and lambda light chains within the glomeruli, tubulointerstitial compartment and arterial vessels. Immunofluorescence, performed on protease digested formalin-fixed paraffin-embedded tissue (IF-P) is shown in Figure 2.
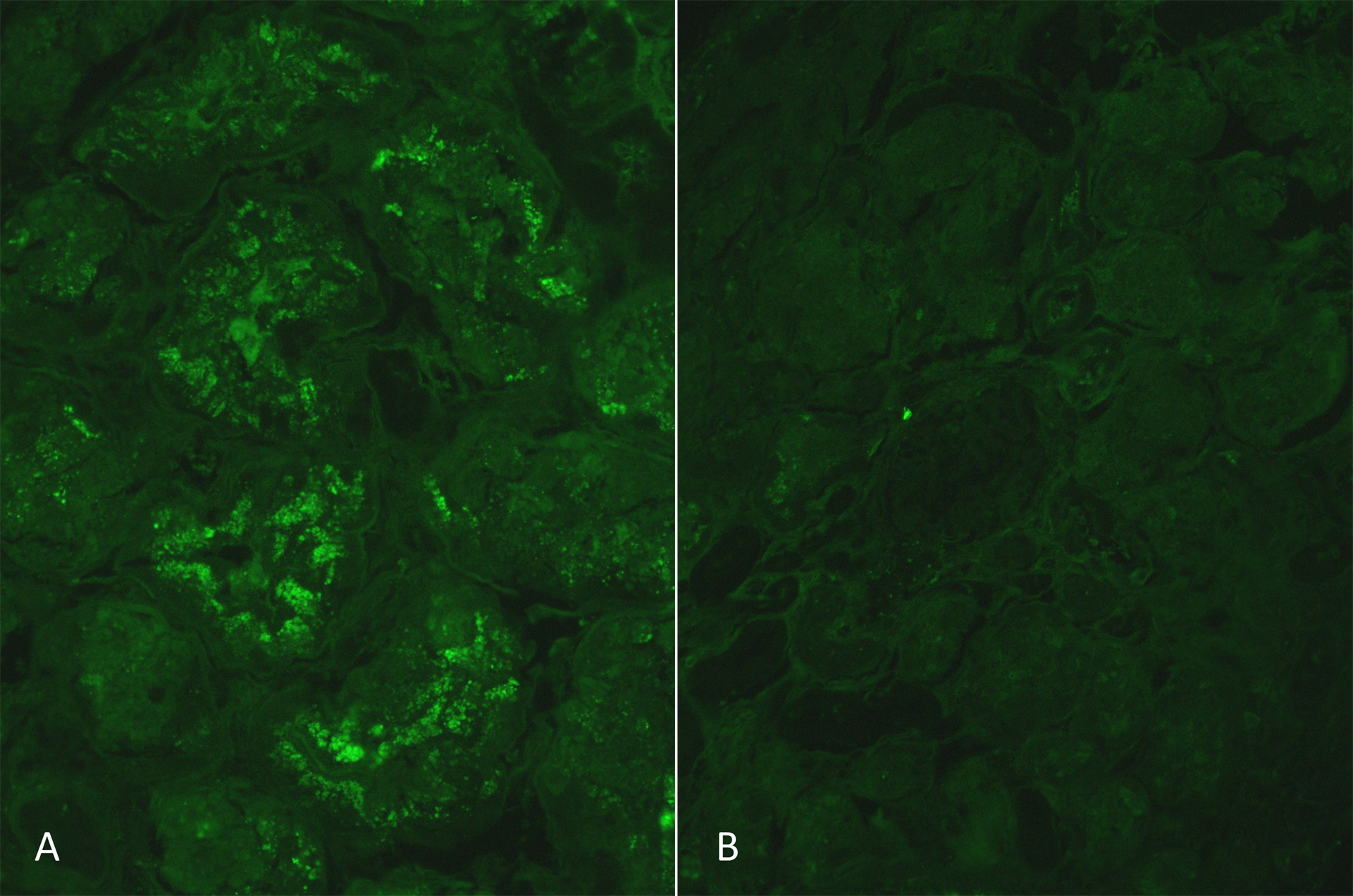
Figure 2: IF-P using FITC-conjugated anti-human kappa and lambda light chains showed a 1-2+ positive staining for kappa light chains mainly within the tubular cells (A). Occasional glomerular epithelial cells positivity was also noted (not shown here). No staining for lambda light chains was noted within the tubular cells (B).
ELECTRON MICROSCOPY (EM):
On EM, podocytes exhibited rare, scattered, round electron dense vacuoles. Foot processes were generally preserved. The endothelial cells showed mild swelling with loss of fenestrations. The capillary basement membranes were normal, with thickness ranging from 300-350nm. The mesangial areas contained focal mild increase in matrix with normal cellularity. No immune complex deposits were identified within the glomerular capillary basement membranes or the mesangial areas. Pertinent tubular findings are summarized in Figure 3.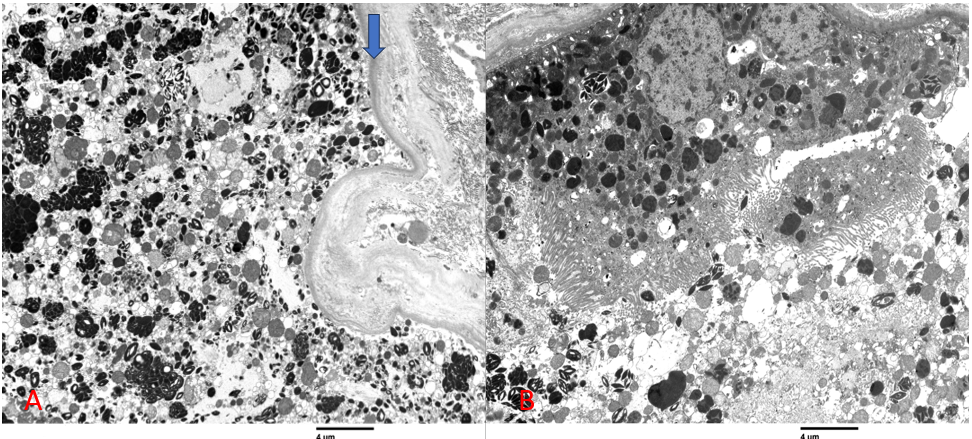
Figure 3. Proximal tubular epithelial cells were variably swollen containing prominent lysosomes with dark stained inclusions (A; arrow marks tubular basement membrane), some of which had crystalline (B) or myelin body configurations. Several crystals can be seen in the tubular lumen (B).
FINAL DIAGNOSIS
- Light chain proximal tubulopathy, mainly non-crystalline and focal crystalline type, associated with monoclonal kappa light chains with occasional glomerular epithelial involvement.
- Focal tubular atrophy with mild interstitial fibrosis, 30% of the cortex.
- Mild global ischemic changes and global glomerulosclerosis.
- No evidence of concomitant monoclonal immunoglobulin deposition disease or Bence Jones cast nephropathy or amyloidosis.
DISCUSSION
Monoclonal gammopathy is defined as detectable monoclonal heavy chain, heavy and light chains or kappa or lambda light chain alone in the circulation. This is as a consequence of an abnormal production of monoclonal immunoglobulins or their components. The source of monoclonal proteins (paraproteins) ranges from indolent B-cell lymphomas to plasma cell neoplasms. The spectrum of clinical presentation also varies – from low-grade entities that do not require treatment (MGUS) to progressive diseases with poor prognosis. These paraproteins can deposit in various organs, causing tissue damage. Pertaining to the kidney, the term monoclonal gammopathy of renal significance (MGRS) was introduced to highlight the evidence of kidney damage related to monoclonal paraproteinemia despite the absence of diagnostic criteria for a hematologic neoplasm [1]. It was shown that renal disease in patients with monoclonal gammopathy or multiple myeloma is associated with worse prognosis [2], which can be improved by timely chemotherapeutic reduction of light chains (LCs) by controlling the neoplastic clone [3].
Renal lesions under the MGRS commonly include AL amyloidosis or monoclonal immunoglobulin deposition disease amongst others, which also involve the tubulo-interstitium and vasculature. Light chain proximal tubulopathy (LCPT) is an uncommon representative of this group, accounting for 4-5% of MGRS (Table 1) [4, 5]. A typical clinical presentation of LCPT is non-nephrotic proteinuria with median protein of 1.5-2.5 g/day or mild CKD (median creatinine of 1.9–2.0 mg/dl) [6]. Complete or incomplete Fanconi’s syndrome is detected in approximately 40% of the cases [4]. Bence Jones proteinuria is almost always present.
Several morphologic forms of LCPT are described, however, it is commonly divided into crystalline and non-crystalline types [5, 7]. Crystalline type is characterized by intracellular crystalline inclusions of a variety of sizes and shapes - rectangular, rhomboid, needle shaped etc. They tend to accumulate in the lysosomes of the proximal tubular epithelium, causing cellular injury and interfering with their function. Monoclonal light chains are freely filtered through the glomerulus and reabsorbed by the proximal tubular epithelial cells via the megalin/cubilin scavenger receptor, followed by their intralysosomal degradation. In LCPT, however, abnormal light chains possess a hydrophobic amino acid side chains due to mutations in the variable domain of the immunoglobulin molecule that confer the ability to resist lysosomal enzyme and protease degradation. Such light-chain crystals or inclusions disrupt lysosomal function, inducing cellular injury, impairing reabsorption, which results in light-chain proximal tubulopathy [8]. More than 90% of the LCs causing LCPT belong to the subgroup 1 of kappa variable domain. In contrast, most cases of lambda LCPT are of non-crystalline type because lambda LCs lack the capability to spontaneously crystallize [9]. Podocytes can sometimes exhibit an accumulation of crystalline inclusions in cases of crystalline type LCPT, leading to nephrotic-range proteinuria [4]. No crystalline podocyte structures were identified in our case. The significance of electron dense podocyte vacuoles that we encountered is unknown. They can either represent a non-specific finding that reflects a low-level proteinuria or be related to the monoclonal protein.
LCPT is a diagnostic challenge provided its rarity and only a fraction of patients having a detectable diagnosis of related hematologic disorder at initial presentation. Only 15% to 50% of the patients had a known MGRS-associated hematologic entity by the time of the renal biopsy [4, 5, 10]. Another challenge is a frequent false-negative light chain immunostaining, when performed on frozen tissue. An antigen retrieval step using enzyme digestion may be required to expose the specific epitopes to achieve a positive staining [11]. The sensitivity of IF-F and IF-P for detection of LCPT was 35% (15/43) and 97% (37/38), respectively. Interestingly, IF-F was significantly better for detecting noncrystalline LCPT than crystalline LCPT [4].
While crystalline and non-crystalline monoclonal light chain deposits can co-exist, crystalline deposits can often be recognized only on ultrastructural examination, with light microscopy showing mostly coarsely granular material in the proximal tubules and failing to show clear crystalline structures [4]. This is demonstrated in the case presented here (see above light and electron microscopy findings). However, a doubt is raised about whether non-crystalline type of LCPT represents a truly independent disease process or is just a physiologic trafficking of light chains [5, 12]. The focal crystal formation in our case does not reflect the extent or severity of tubular damage that is seen by light microscopy and corroborated by the laboratory data. This suggests that non-crystalline component of LCPT is accountable for much of the proximal tubular epithelial dysfunction leading to acute kidney injury (AKI).
The mainstay of MGRS treatment is controlling the neoplastic clone with chemotherapy or stem cell transplantation - approaches that were earlier utilized only in patients with an established diagnosis of hematologic malignancy. Free light chain reduction in patients with monoclonal gammopathies is associated with better renal recovery, resulting in improved patient survival [13].
|
Epidemiology and renal manifestation |
Light microscopy |
Immunofluorescence |
Electron microscopy |
|
|
Light chain cast nephropathy |
33% of patients with multiple myeloma [14]. AKI (median eGFR: 13 mL/min/1.73 m2), non-albumin proteinuria (median 24-h urine protein: 2.2 g and median urinary albumin excretion: 9%), progressive CKD. |
Brightly eosinophilic, pale pink on PAS, fuscinophilic on Trichrome stain casts in distal segments of nephron. Casts have sharp angulated borders, fractured appearance. tubular epithelial injury, tubulo-interstitial inflammation, and focal giant cell reaction. |
Monoclonal light chain restriction (either kappa or lambda), variable amounts of uromodulin deposits by IHC, pronase digestion retrieval is occasionally necessary to unmask the monotypic composition of tubular casts. |
Mostly non-specific granular electron-dense material with occasional crystalline substructures. |
|
AL amyloidosis |
21% in patients with multiple myeloma [14]. Heavy proteinuria (median protein, 5–6 g/day), nephrotic syndrome (66%), mild CKD (median creatinine, 1.2 mg/dl). |
Hypocellular, amorphous, mesangial and capillary wall deposits (spicular type), pale staining with PAS, PASM, Congo red positive with pale green birefringence (polarized), variable TBM, rarely in casts, interstitial and arterial wall deposits. |
Pseudolinear to smudgy staining for monotypic lambda > kappa (7:1) light chain or heavy chain within the amyloid deposits. |
Randomly oriented, rigid, non-branching fibrils, solid cross section, replacing the extracellular matrix, ranging 8-12 nm in diameter |
|
LCPT |
4-5% of other dysproteinemia-related renal diseases. Proteinuria (median protein, 1.5– 2.5 g/day), mild CKD (median creatinine, 1.9–2.0 mg/dl), proximal tubulopathy with or without complete Fanconi’s syndrome |
Varying degrees of tubular cell injury, loss of brush borders, ballooning or cytoplasmic pallor giving a feathery appearance by H&E, PAS-negative and fuscinophilic, cytoplasmic or lysosomal granular deposits (non-crystalline type). Variously shaped PAS-positive, fuscinophilic crystalline deposits (crystalline type). |
Commonly monoclonal kappa light chain staining (crystalline type). May require pronase digestion retrieval on paraffin-embedded tissue sections to confirm diagnosis. |
Tubular cytoplasmic or intralysosomal amorphous, crystalline (rod, rhomboid or needle shaped) or tubulo-fibrillar deposits seen, nearly 1/3 of non-crystalline lysosomal inclusions are lambda light chain positive. |
|
Crystal-storing histiocytosis |
Rare, with ~20 renal cases described in the literature [15]. Can sometimes occur in combination with other MGRS-causing conditions [16] Proteinuria (24-h urine protein range: 2.4-4.6 g), reduced kidney function (serum creatinine range: 2.38-5.01 mg/dL) |
Interstitial and glomerular histiocytes containing hyper-eosinophilic crystalline inclusions, pseudo-Gaucher cells |
Positive staining for Ig fragments (most commonly IgG kappa LC), positive CD68 IHC stain confirms the histiocytic phenotype of the cells containing the crystalline inclusions |
Electron-dense needle-shaped crystals within the histiocytes in the interstitium and glomerular capillaries |
|
Light-chain-mediated acute tubular interstitial nephritis (Tubulopathy Associated With Interstitial Inflammatory Reaction) [7] |
22% of the monoclonal dysproteinemia-related kidney biopsies (28/126) in one study. AKI or CKD, non-nephrotic range proteinuria. |
Interstitial inflammation composed of lymphocytes and scattered plasma cells with occasional eosinophils. Tubular epithelial damage with occasional tubulitis. |
Granular monoclonal light-chain staining in the cytoplasm of proximal tubular cells with accompanying linear staining along tubular basement membranes in the areas of inflammation. 16/28 cases with kappa and 12/28 cases with lambda light chain-restricted deposits. |
Tubules with prominent cytoplasmic lysosomes and evidence of tubular damage, generally associated with areas with inflammation and tubulitis. In 7 cases, there was a focal deposition of punctate electron-dense material along tubular basement membranes. |
Table 1. A summary of common MGRS as well as some non-MGRS related renal lesions primarity affecting the tubulo-interstitium (adapted with modifications from [6] and [9]).
References
- Leung N, Bridoux F, Batuman V, et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group [published correction appears in Nat Rev Nephrol. 2019 Feb;15(2):121]. Nat Rev Nephrol. 2019;15(1):45-59. doi:10.1038/s41581-018-0077-4
- Stratta P, Gravellone L, Cena T, Rossi D, Gaidano G, Fenoglio R, et al. Renal outcome and monoclonal immunoglobulin deposition disease in 289 old patients with blood cell dyscrasias: a single center experience. Crit Rev Oncol Hematol. (2011) 79:31–42. 10.1016/j.critrevonc.2010.05.001
- Sayed RH, Wechalekar AD, Gilbertson JA, Bass P, Mahmood S, Sachchithanantham S, et al. Natural history and outcome of light chain deposition disease. Blood. (2015) 126:2805–10. 10.1182/blood-2015-07-658872
- Stokes MB, Valeri AM, Herlitz L, Khan AM, Siegel DS, Markowitz GS, D'Agati VD. Light Chain Proximal Tubulopathy: Clinical and Pathologic Characteristics in the Modern Treatment Era. J Am Soc Nephrol. 2016 May;27(5):1555-65. doi: 10.1681/ASN.2015020185. Epub 2015 Sep 15. PMID: 26374607; PMCID: PMC4849818.
- Larsen CP, Bell JM, Harris AA, Messias NC, Wang YH, Walker PD. The morphologic spectrum and clinical significance of light chain proximal tubulopathy with and without crystal formation. Mod Pathol. 2011;24(11):1462-1469. doi:10.1038/modpathol.2011.104
- Leung N, Bridoux F, Nasr SH. Monoclonal Gammopathy of Renal Significance. N Engl J Med. 2021;384(20):1931-1941. doi:10.1056/NEJMra1810907
- Herrera GA. Proximal tubulopathies associated with monoclonal light chains: the spectrum of clinicopathologic manifestations and molecular pathogenesis. Arch Pathol Lab Med. 2014;138(10):1365-1380. doi:10.5858/arpa.2013-0493-OA
- Luciani A, Sirac C, Terryn S, et al. Impaired lysosomal function underlies monoclonal light chain-associated renal Fanconi syndrome. J Am Soc Nephrol 2016;27:2049-2061
- Sy-Go JPT, Herrmann SM, Seshan SV. Monoclonal Gammopathy-Related Kidney Diseases. Adv Chronic Kidney Dis. 2022;29(2):86-102.e1. doi:10.1053/j.ackd.2022.01.004
- Kousios A, Blakey S, Moran L, et al. Non-crystalline light chain proximal tubulopathy, a morphologically protean entity [published online ahead of print, 2023 Apr 29]. Nephrol Dial Transplant. 2023;gfad085. doi:10.1093/ndt/gfad085
- Nasr SH, Fidler ME, Said SM. Paraffin Immunofluorescence: A Valuable Ancillary Technique in Renal Pathology. Kidney Int Rep. 2018 Jul 7;3(6):1260-1266. doi: 10.1016/j.ekir.2018.07.008. PMID: 30450452; PMCID: PMC6224795
- Büttner-Herold M, Krieglstein N, Chuva T, Minuth K, Pfister F, Daniel C, Klewer M, Büttner A, Ferrazzi F, Bertz S, Amann K. Light Chain Restriction in Proximal Tubules-Implications for Light Chain Proximal Tubulopathy. Front Med (Lausanne). 2022 Mar 28;9:723758. doi: 10.3389/fmed.2022.723758. PMID: 35419374; PMCID: PMC8995435.
- Hutchison CA, Heyne N, Airia P, Schindler R, Zickler D, Cook M, et al. Immunoglobulin free light chain levels and recovery from myeloma kidney on treatment with chemotherapy and high cut-off haemodialysis. Nephrol Dial Transplant. (2012) 27:3823–8. 10.1093/ndt/gfr773
- Nasr SH, Valeri AM, Sethi S, et al. Clinicopathologic correlations in multiple myeloma: a case series of 190 patients with kidney biopsies. Am J Kidney Dis. 2012;59(6):786-794. doi:10.1053/j.ajkd.2011.12.028
- Gupta RK, Rosenberg AZ, Bagnasco SM, Arend LJ. Renal crystal-storing histiocytosis involving glomeruli - A comprehensive clinicopathologic analysis. Ann Diagn Pathol. 2019;43:151403. doi:10.1016/j.anndiagpath.2019.151403
- Zhu L, Wang L, Shi H, et al. Combined crystal-storing histiocytosis, light chain proximal tubulopathy, and light chain crystalline podocytopathy in a patient with multiple myeloma: a case report and literature review. Ren Fail. 2023;45(1):2145970. doi:10.1080/0886022X.2022.2145970
About RPS
The RPS promotes excellence in diagnosis, fosters basic, clinical and translational research, encourages training and education in renal disease, sponsors US based and international conferences and symposia, and brings news and updates pertaining to renal pathology to its members around the world.  | ContactsOffice of the Secretary Mei Lin Z. Bissonnette MD PhD Office of the Treasurer Kuang-Yu Jen, MD, PhD |
Copyright Renal Pathology Society © 2018. - Privacy Policy Terms & Conditions Cookie Policy