RPS Caso del Mes
Dic 2023
Hallazgo inesperado en un hombre de 65 años con enfermedad renal crónica y proteinuria leve
Siarhei Dzedzik1
Ilya Glezerman1,2
Surya V. Seshan1
1. New York-Presbyterian/Weill Cornell Medicine, New York, NY2. Memorial Sloan Kettering Cancer Center, New York, NY
CASO CLÍNICO
Un hombre de 65 años buscó atención médica en un hospital local después de una caída. Estudios radiográficos revelaron una lesión osteolítica con sospecha de fractura patológica en la vertebra C6. Se reseccionó la lesión seguido por radioterapia. La patología mostró células tumorales positivas para CD138 con restricción de kappa, consistente con el diagnóstico de plasmocitoma solitario. Dos años y medio después, el paciente fue evaluado por aumento de creatinina (de 1.0 a 1.5 mg/dL). El examen físico y el panel metabólico completo fueron normales. Sin embargo, hubo un aumento significativo en las cadenas ligeras kappa a 38.09 (0.33-1.94) mg/dL, una proporción de cadenas kappa/lambda de 35.93 (0.26-1.65). La electroforesis de proteínas en sangre fue negativa para el pico M, aunque la prueba de inmunofijación en sangre fue positiva para IgA kappa. La electroforesis de proteínas en orina fue negativa para el pico M, aunque la inmunofijación fue positiva para las cadenas ligeras kappa. El examen de orina no mostró células ni precipitaciones renales, y la relación proteína creatinina en orina fue inferior a 0.1. Diez meses después, se realizó una biopsia de riñón porque hubo elevación de las cadenas ligeras kappa (84.07 mg/dL) y de la proteinuria (relación proteína/creatinina en orina de 0.6).
BIOPSIA RENAL
La microscopia óptica reveló leves cambios isquémicos en nueve de los cuarenta y siete glomérulos (9/47). Los glomérulos restantes no mostraron cambios significativos. El 30% del tejido cortical demostraba atrofia tubular con fibrosis intersticial leve, e infiltración inflamatoria intersticial crónico mínimos (linfocitos mezclados con macrófagos y células plasmáticas dispersas. Los hallazgos tubulares se resumen en la Figura 1. 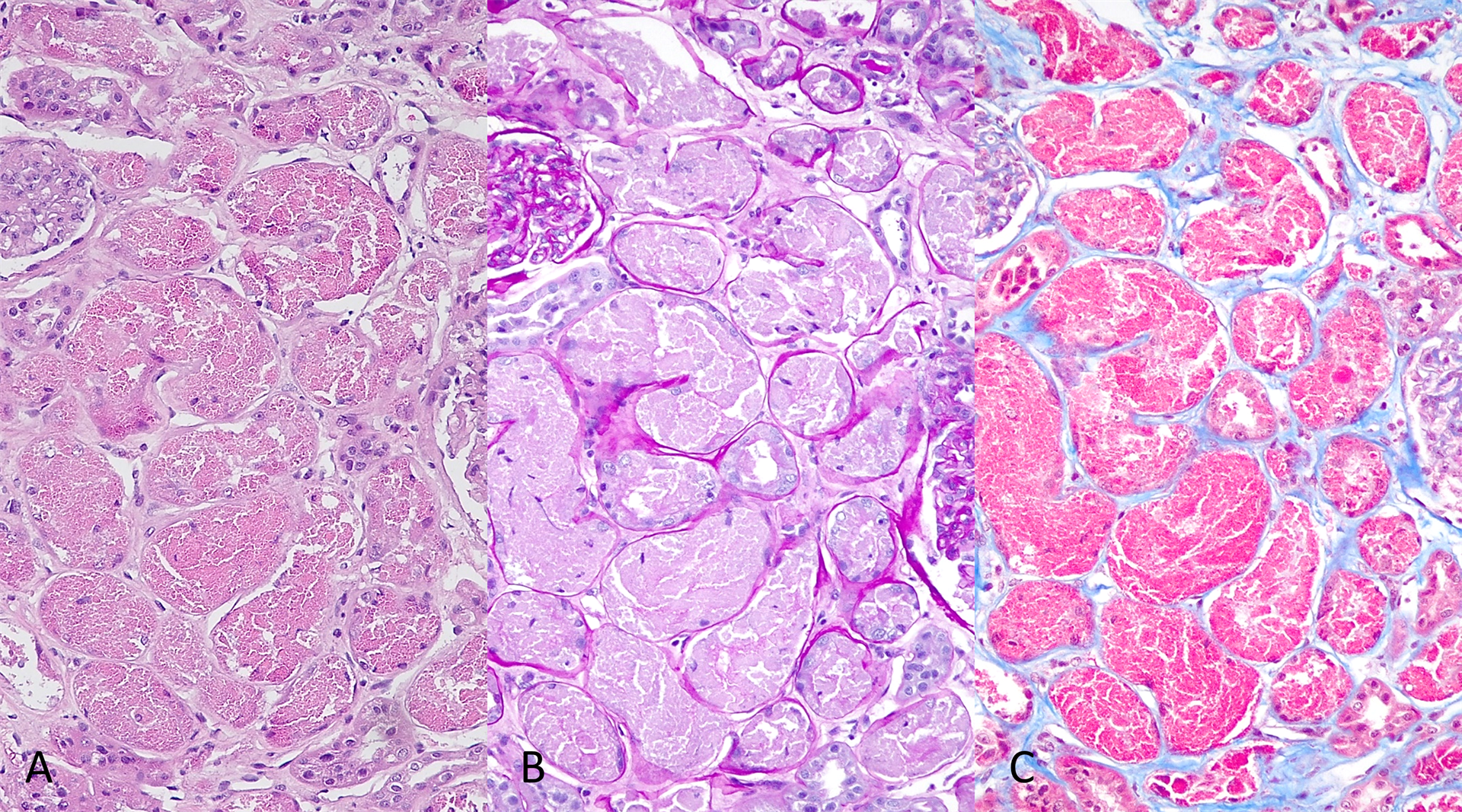
Figura 1: La mayoría de los túbulos proximales están dilatados. Las células epiteliales están distendidas por material grueso y eosinofílico (A) que es negativo en el PAS (B) y fuertemente fucsinafílico en el tricrómico de Masson (C). Hay atenuación del borde en cepillo, picnosis, pérdida de cohesión epitelial y desprendimiento de las células epiteliales en el lumen tubular.

INMUNOFLUORESCENCIA:
La IF (2 glomérulos), realizada en tejido congelado (IF-F), fue negativa para IgG, IgA, C3, C1q, kappa y lambda en los glomérulos, en el compartimento tubulointersticial y vasos arteriales. La IF en parafina digerida por proteasa (IF-P) se muestra en la Figura 2.
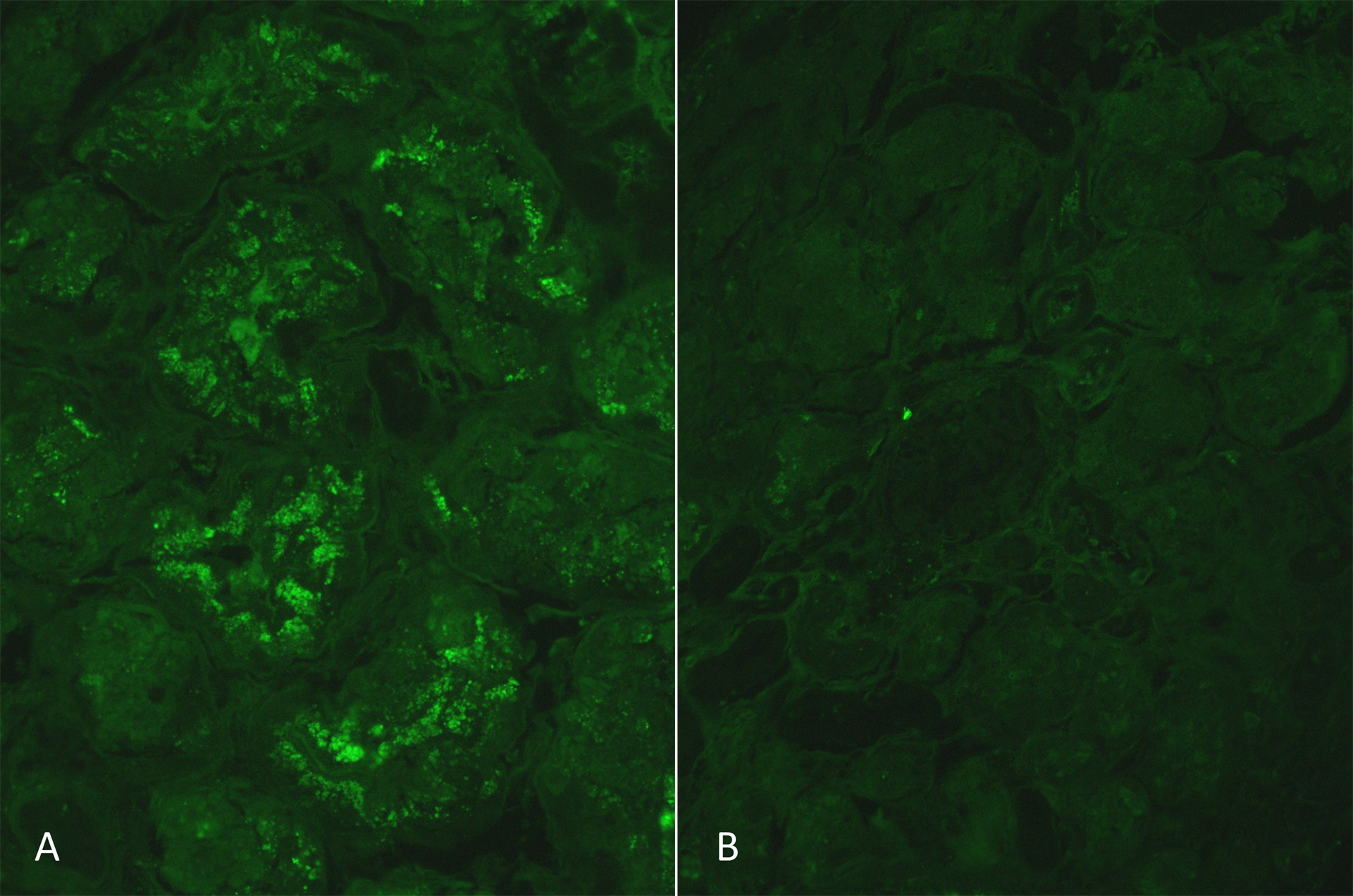
Figura 2: IF-P anti-cadenas ligeras kappa y lambda humanas fue positiva 1-2+ para cadenas kappa en las células epiteliales tubulares (A) y ocasional positividad en las células epiteliales glomerulares (no mostradas). No se observó positividad para las cadenas ligeras lambda en las células epiteliales tubulares (B).
MICROSCOPIA ELECTRÓNICA:
Los podocitos mostraron vacuolas electrodensas redondeadas raras, y los procesos podocitarios estaban preservados. Las células endoteliales presentaban leve hinchazón y pérdida de fenestraciones. Las membranas basales capilares estaban normales, con un grosor entre 300-350 nm. Las áreas mesangiales contenían un leve aumento focal de la matriz con celularidad normal. No se identificaron depósitos de complejos inmunes en las membranas basales capilares glomerulares o en las áreas mesangiales. Los hallazgos tubulares se resumen en la Figura 3.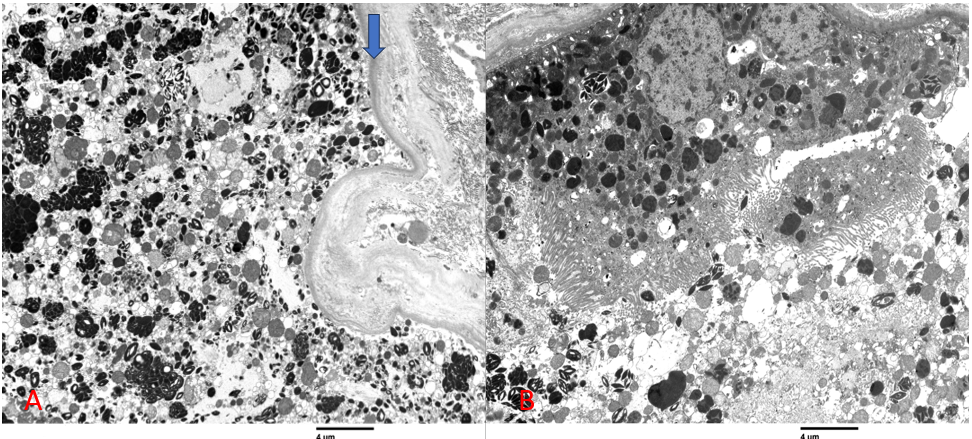
Figura 3. Las células epiteliales tubulares proximales estaban variadamente hinchadas, con lisosomas prominentes con inclusiones oscuras (A; la flecha azul indica la membrana basal tubular), algunas de las cuales tenían configuraciones cristalinas (B) o en forma de cuerpo de mielina. Se pueden ver cristales en el lumen tubular (B).
DIAGNÓSTICO FINAL
- Tubulopatía proximal por cadenas ligeras, principalmente del tipo no cristalino, con cristales focales, asociada con cadenas ligeras kappas monoclonales y ocasional afectación epitelial glomerular.
- Atrofia tubular focal con fibrosis intersticial, afectando al 30% del córtex.
- Cambios isquémicos leves y glomeruloesclerosis global focal.
- Ausencia de enfermedad por depósito de inmunoglobulinas monoclonales, nefropatía por cilindros o amiloidosis renal.
DISCUSIÓN
La gammapatía monoclonal es el resultado de cadenas pesadas monoclonales circulantes, cadenas pesadas y ligeras monoclonales o cadenas ligeras (kappa o lambda). El origen de las proteínas monoclonales (paraproteínas) varía desde linfomas de células B indolentes hasta neoplasias de células plasmáticas. El espectro de presentación clínica también varía, desde entidades de bajo grado que no requieren tratamiento (gammapatía monoclonal de significado incierto, GMSI) hasta enfermedades progresivas con mal pronóstico. Estas paraproteínas pueden depositarse en varios órganos, causando daño a los tejidos. Las gammpatías monoclonales de significado renal (GMSR) abarcan enfermedades caracterizadas por daño renal relacionado con paraproteinemia monoclonal, a pesar de la ausencia de criterios diagnósticos para una neoplasia hematológica [1]. Se ha demostrado que la enfermedad renal en pacientes con gammapatía monoclonal o mieloma múltiple se asocia con un peor pronóstico [2], que puede mejorarse mediante el control oportuno de la reducción de cadenas ligeras a través de una terapia dirigid a clones de células plasmáticas.
Las lesiones renales de GMSR comúnmente incluyen amiloidosis AL, enfermedad de depósito de inmunoglobulina monoclonal, entre otras, que también afectan el túbulo-intersticio y la vasculatura. La tubulopatía proximal por cadenas ligeras (TPCL) es una representante rara de este grupo, correspondiendo al 4-5% de GMSR (Tabla 1) [4, 5]. Una presentación clínica típica de la TPCL es la proteinuria subnefrótica (media 1,5-2,5 g/día) o enfermedad renal crónica leve (creatinina media 1,9-2,0 mg/dL) [6]. El síndrome de Fanconi completo o incompleto se detecta en aproximadamente el 40% de los casos [4] y la proteinuria de Bence Jones está casi siempre presente.
A pesar de tener varias variantes morfológicas, el TPCL se divide comúnmente en subtipos cristalinos y no cristalinos [5, 7]. El tipo cristalino se caracteriza por inclusiones cristalinas intracelulares de varios tamaños y formas: rectangulares, romboides, en forma de aguja, etc. Ellos se acumulan en los lisosomas del epitelio del túbulo proximal, causando lesiones celulares e interfiriendo en su función. Las cadenas ligeras monoclonales son filtradas por el glomérulo y reabsorbidas por las células epiteliales tubulares proximales a través del receptor scavenger megalina/cubilina, seguido de su degradación lisosomal. En la TPCL, las cadenas ligeras anormales tienen cadenas laterales de aminoácidos hidrofóbicos debido a mutaciones en el dominio variable de la molécula de inmunoglobulina, lo que les confiere la capacidad de resistir la degradación lisosomal. Estos cristales de cadenas ligeras interfieren en la función lisosómica, induciendo lesiones celulares y perjudicando la reabsorción tubular proximal [8]. Más del 90% de las cadenas ligeras que causan TPCL pertenecen al subgrupo 1 del dominio variable kappa. En contraste, la mayoría de los casos de TPCL lambda son del tipo no cristalino porque las cadenas ligeras lambda no tienen la capacidad de cristalizar espontáneamente [9].
Podocytes can sometimes exhibit an accumulation of crystalline inclusions in cases of crystalline type LCPT leading to nephrotic-range proteinuria [4]. Nocrystalline podocyte structures were identified in our case. The significance of electron dense podocyte vacuoles that we encountered is unknown. They can either represent a non-specific finding that reflects a low-level proteinuria or be related to the monoclonal protein. La TPCL es un desafío diagnóstico debido a su rareza. Además, sólo algunos pacientes tienen un trastorno hematológico previamente diagnosticado en el momento de la presentación. Las cohortes anteriores muestran que el 15% al 50% de los pacientes tenían una entidad hematológica relacionada con el GMSR conocida en el momento de la biopsia renal [4, 5, 10]. La alta tasa de falsos negativos en la IF para cadenas ligeras en tejido congelado es otro obstáculo para un diagnóstico preciso de la TPCL. Puede ser necesario la recuperación de antígeno mediante la digestión enzimática para exponer los epítopos específicos y obtener una IF positiva [11]. La sensibilidad de la prueba de inmunofluorescencia por congelación (IF-F) y la técnica en tejidos preservados (IF-P) fue del 35% (15/43) y el 97% (37/38), respectivamente, para el diagnóstico de TPCL. Curiosamente, la IF-F fue significativamente mejor para detectar TPCL no cristalina que TPCL cristalina [4].
Aunque los depósitos de cadenas ligeras monoclonales cristalinos y no cristalinos pueden coexistir, normalmente se necesita un examen ultraestructural para identificar los depósitos cristalinos [4], como en el caso presente. Sin embargo, no se sabe si el TPCL representa una enfermedad verdaderamente independiente o es simplemente el resultado del tráfico fisiológico de cadenas ligeras [5, 12]. La formación focal de cristales en nuestro caso no refleja la extensión o gravedad de la lesión tubular vista en la microscopía óptica y respaldada por los datos de laboratorio. Esto sugiere que el componente no cristalino de la TPCL es responsable por la disfunción epitelial tubular proximal que conduce a la lesión renal aguda (LRA).
El clavel del tratamiento del GMSR es el control del clon de células plasmáticas con quimioterapia o trasplante de células madre. La reducción de las cadenas ligeras libres en pacientes con gammapatía monoclonales se asocia con una mejor recuperación renal y supervivencia del paciente [13].
|
Epidemiología y manifestación renal |
Microscopía óptica |
Inmunofluorescencia |
Microscopía electrónica |
|
|
Nefropatía por cilindros |
33% de los pacientes con mieloma múltiple [14]. IRA (TFG medio: 13 ml/min/1,73 m2), proteinuria (media 24 h: 2,2 g; excreción media de albúmina: 9%), ERC progresiva. |
Cristales eosinófilos, rosa pálido en PAS, fuscinófilos en tricrómico en segmentos distales de nefronas. Los cristales tienen bordes angulados afilados y apariencia fracturada. Lesión epitelial tubular, inflamación tubulointersticial y reacción focal de células gigantes. |
Restricción de cadenas ligeras monoclonales (kappa o lambda), cantidades variables de depósitos de uromodulina por IHQ (inmunohistoquímica), ocasionalmente, es necesario el uso de recuperación con digestión con pronasa para revelar la composición monotípica de los cilindros tubulares. |
Material granular electrónicamente denso, en su mayoría inespecífico, con subestructuras cristalinas ocasionales. |
|
Amiloidosis AL |
21% en pacientes con mieloma múltiple [14]. Proteinuria masiva (media 5-6 g/día), síndrome nefrótico (66%), ERC moderada (creatinina media, 1,2 mg/dl). |
Depósitos hipocelulares amorfos en paredes mesangiales y capilares (tipo espicular), pálido en el PAS, PASM, positivo en el rojo Congo y birrefringencia verde pálida (polarizada), membrana basal tubular variable, raro en cilindros, intersticio y paredes arteriales. |
Tinción pseudolineal a borrosa para cadena ligera o pesada monotípica lambda > kappa (7:1) dentro de los depósitos de amiloide. |
Fibrillas rígidas, no ramificadas, orientadas aleatoriamente, de sección transversal sólida, que reemplazan la matriz extracelular, con un diámetro de entre 8 y 12 nm. |
|
TPCL |
El 4-5% de otras enfermedades renales relacionadas con paraproteínas. Proteinuria (proteína media, 1,5-2,5 g/día), enfermedad renal crónica leve (creatinina media, 1,9-2,0 mg/dl), tubulopatía proximal con o sin síndrome de Fanconi. |
Diversos grados de daño de las células tubulares, pérdida de los bordes en cepillo, hinchazón o palidez citoplasmática, dando una apariencia borrosa en H&E, negativo en PAS y fuscinófilo, con depósitos granulares citoplasmáticos o lisosomales (tipo no cristalino). Depósitos cristalinos fuscinófilos (similares a cristalinos), PAS positivos, de forma variable. |
Tinción monoclonal de cadena ligera Kappa (tipo cristalino). Se requiere recuperación con digestión con pronasa para confirmar el diagnóstico. |
Se observan depósitos tubulares citoplasmáticos o intralisosomales amorfos, cristalinos (rómbicos o en forma de agujas) o fibrilares. Aproximadamente 1/3 de las inclusiones lisosomales no cristalinas son positivas para cadena ligera lambda. |
|
Histiocitosis por almacenamiento de cristales (HAC) |
Raro, con aproximadamente 20 casos renales descritos en la literatura [15]. Puede ocurrir en combinación con otras condiciones que causan GMSR [16]. Proteinuria (rango de proteínas en orina de 24 horas: 2,4-4,6 g), función renal reducida (rango de creatinina sérica: 2,38-5,01 mg/dL). |
Histiocitos intersticiales y glomerulares que contienen inclusiones cristalinas hipereosinófilas, células pseudo-Gaucher. |
La tinción positiva para fragmentos de Ig (más comúnmente cadena leve IgG kappa). CD68 confirma el fenotipo histiocítico de las células que contienen inclusiones cristalinas. |
Cristales electrodensos en forma de aguja en histiocitos intersticiales y capilares glomerulares. |
|
Nefritis intersticial aguda asociada a cadenas ligeras (tubulopatía asociada a reacción inflamatoria intersticial) [7] |
El 22% de las biopsias renales se relacionaron con disproteinemia monoclonal (28/126). IRA o ERC con proteinuria subnefrótica. |
Inflamación intersticial con linfocitos, células plasmáticas y eosinófilos ocasionales. Daño epitelial tubular con tubulitis. |
Tinción granular de cadenas ligeras monoclonales en el citoplasma de las células tubulares proximales; tinción lineal a lo largo de las membranas basales tubulares en áreas de inflamación. 16/28 casos con cadenas ligeras kappa y 12/28 casos con cadenas ligeras lambda. |
Túbulos con lisosomas prominentes, áreas de inflamación y tubulitis. En 7 casos, hubo un depósito focal granular eléctron-denso a lo largo de las membranas basales tubulares. |
Tabla 1. Lesiones renales relacionadas con GMSR y algunas no relacionadas con GMSR que afectan principalmente al túbulo-intersticio (adaptado con modificaciones de [6] y [9]).
Referencias
- Leung N, Bridoux F, Batuman V, et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group [published correction appears in Nat Rev Nephrol. 2019 Feb;15(2):121]. Nat Rev Nephrol. 2019;15(1):45-59. doi:10.1038/s41581-018-0077-4
- Stratta P, Gravellone L, Cena T, Rossi D, Gaidano G, Fenoglio R, et al. Renal outcome and monoclonal immunoglobulin deposition disease in 289 old patients with blood cell dyscrasias: a single center experience. Crit Rev Oncol Hematol. (2011) 79:31–42. 10.1016/j.critrevonc.2010.05.001
- Sayed RH, Wechalekar AD, Gilbertson JA, Bass P, Mahmood S, Sachchithanantham S, et al. Natural history and outcome of light chain deposition disease. Blood. (2015) 126:2805–10. 10.1182/blood-2015-07-658872
- Stokes MB, Valeri AM, Herlitz L, Khan AM, Siegel DS, Markowitz GS, D'Agati VD. Light Chain Proximal Tubulopathy: Clinical and Pathologic Characteristics in the Modern Treatment Era. J Am Soc Nephrol. 2016 May;27(5):1555-65. doi: 10.1681/ASN.2015020185. Epub 2015 Sep 15. PMID: 26374607; PMCID: PMC4849818.
- Larsen CP, Bell JM, Harris AA, Messias NC, Wang YH, Walker PD. The morphologic spectrum and clinical significance of light chain proximal tubulopathy with and without crystal formation. Mod Pathol. 2011;24(11):1462-1469. doi:10.1038/modpathol.2011.104
- Leung N, Bridoux F, Nasr SH. Monoclonal Gammopathy of Renal Significance. N Engl J Med. 2021;384(20):1931-1941. doi:10.1056/NEJMra1810907
- Herrera GA. Proximal tubulopathies associated with monoclonal light chains: the spectrum of clinicopathologic manifestations and molecular pathogenesis. Arch Pathol Lab Med. 2014;138(10):1365-1380. doi:10.5858/arpa.2013-0493-OA
- Luciani A, Sirac C, Terryn S, et al. Impaired lysosomal function underlies monoclonal light chain-associated renal Fanconi syndrome. J Am Soc Nephrol 2016;27:2049-2061
- Sy-Go JPT, Herrmann SM, Seshan SV. Monoclonal Gammopathy-Related Kidney Diseases. Adv Chronic Kidney Dis. 2022;29(2):86-102.e1. doi:10.1053/j.ackd.2022.01.004
- Kousios A, Blakey S, Moran L, et al. Non-crystalline light chain proximal tubulopathy, a morphologically protean entity [published online ahead of print, 2023 Apr 29]. Nephrol Dial Transplant. 2023;gfad085. doi:10.1093/ndt/gfad085
- Nasr SH, Fidler ME, Said SM. Paraffin Immunofluorescence: A Valuable Ancillary Technique in Renal Pathology. Kidney Int Rep. 2018 Jul 7;3(6):1260-1266. doi: 10.1016/j.ekir.2018.07.008. PMID: 30450452; PMCID: PMC6224795
- Büttner-Herold M, Krieglstein N, Chuva T, Minuth K, Pfister F, Daniel C, Klewer M, Büttner A, Ferrazzi F, Bertz S, Amann K. Light Chain Restriction in Proximal Tubules-Implications for Light Chain Proximal Tubulopathy. Front Med (Lausanne). 2022 Mar 28;9:723758. doi: 10.3389/fmed.2022.723758. PMID: 35419374; PMCID: PMC8995435.
- Hutchison CA, Heyne N, Airia P, Schindler R, Zickler D, Cook M, et al. Immunoglobulin free light chain levels and recovery from myeloma kidney on treatment with chemotherapy and high cut-off haemodialysis. Nephrol Dial Transplant. (2012) 27:3823–8. 10.1093/ndt/gfr773
- Nasr SH, Valeri AM, Sethi S, et al. Clinicopathologic correlations in multiple myeloma: a case series of 190 patients with kidney biopsies. Am J Kidney Dis. 2012;59(6):786-794. doi:10.1053/j.ajkd.2011.12.028
- Gupta RK, Rosenberg AZ, Bagnasco SM, Arend LJ. Renal crystal-storing histiocytosis involving glomeruli - A comprehensive clinicopathologic analysis. Ann Diagn Pathol. 2019;43:151403. doi:10.1016/j.anndiagpath.2019.151403
- Zhu L, Wang L, Shi H, et al. Combined crystal-storing histiocytosis, light chain proximal tubulopathy, and light chain crystalline podocytopathy in a patient with multiple myeloma: a case report and literature review. Ren Fail. 2023;45(1):2145970. doi:10.1080/0886022X.2022.2145970
About RPS
The RPS promotes excellence in diagnosis, fosters basic, clinical and translational research, encourages training and education in renal disease, sponsors US based and international conferences and symposia, and brings news and updates pertaining to renal pathology to its members around the world.  | ContactsOffice of the Secretary Mei Lin Z. Bissonnette MD PhD Office of the Treasurer Kuang-Yu Jen, MD, PhD |
Copyright Renal Pathology Society © 2018. - Privacy Policy Terms & Conditions Cookie Policy